IEC 60601-1 Medical Design Standards for Power Supplies

Standards are an integral part of product design and development, and are certainly important in medical applications. While some technical standards — such as IEEE 802 for Wi-Fi — only define final performance, standards for medical design have evolved in recent years to go much deeper, covering design methodology and verification, safety and risk assessment, implementation, and much more.
In this paper we will look at the IEC 60601-1 medical standard and its impact on power supply design. IEC 60601-1 provides general requirements, in a series of standards, that address the basic safety and essential performance requirements of medical electrical equipment. We will see how the standard has evolved, through to the 4th edition EMC collateral standard that establishes new “environments” when considering electromagnetic compatibility (EMC) between medical electrical equipment and other electronic devices.
IEC 60601-1 OVERVIEW
What is IEC 60601-1?
IEC 60601 is a series of technical standards for the safety and effectiveness of medical electrical equipment.
The Primary Standard
The primary standard governing medical device design is formally known as IEC 60601-1 - Medical electrical equipment - Part 1: General requirements for basic safety and essential performance. More simply it is referred to as IEC 60601-1 or just “60601,” and compliance with this standard has become a de facto requirement for bringing new medical devices to market in many countries. We will look at the global adoption of the standard in more detail later, but it is worth noting that there are European (EN 60601-1) and Canadian (CSA 60601-1) versions of the standard that are identical to the IEC standard. There are also deviations from the standard that relate to country-specific requirements.
Collateral Standards
Within IEC 60601-1, there are “collateral” standards that are denoted as IEC 60601-1-x; for example, IEC 60601-1-2 is the EMC collateral standard mentioned above. Other collateral standards include 60601-1-3, covering radiation protection for diagnostic x-ray systems, 60601-1-9 relating to environmental design, and 60601-1-11 recently introduced for home healthcare equipment.
Particular Standards
As well as collateral standards, there are also many “particular” standards, denoted as IEC 60601-2-x that define specific requirements related to particular types of products, e.g. 60601-2-16 covers blood dialysis and filtration equipment. These particular standards are largely outside the scope of this paper.
The Evolution of IEC 60601
The IEC 60601 standard has a long history with a number of revisions. The original IEC 60601-1 for medical devices was published in 1977. The 2nd edition, published in 1988, focused on safety within the vicinity of a patient. In 2005, the IEC released the 3rd edition, which reflected a further change of perspective, looking at “means of protection” (MOP) both for patients and equipment operators. A substantial amendment to the 3rd edition, known as Edition 3.1, was introduced in 2012. This addressed numerous ambiguities arising from evolving medical equipment technology. Most recently, in 2014, the 4th edition of collateral standard IEC 60601-1-2, “Electromagnetic disturbances – Requirements and tests,” was published.
IEC 60601-1 AND ITS RELEVANCE TO POWER SUPPLIES
The IEC 60601-1 standard has a significant impact on the product development process, going beyond performance test and verification. This is because product complexity generally yields innumerable potential test cases, permutations, and combinations in both normal and abnormal operating modes, and these cannot be assessed in the final design alone.
While power supplies by themselves are not medical devices and, are therefore, not directly covered by the IEC 60606-1 directive, they are nevertheless integral to the design and operation of medical equipment.

2nd Edition - Patient Focus
The intrinsic nature of an ac-dc power supply or a dc-dc converter means the voltages and currents they provide can potentially be hazardous if not properly managed. In the context of safe operation of medical equipment, the 2nd edition of IEC 60601-1 established risk guidelines that applied when a device was within a 6-foot radius from the patient, referred to as the “patient vicinity.” Three use categories of increasing severity were defined:
This classification determined applicable levels of isolation, insulation, creepage, clearance, and leakage as highlighted in figure 2.
| 2nd Edition Requirements by Classification | |||
|---|---|---|---|
| Classification | Isolation | Creepage | Insulation |
| Type B | 1500 Vac | 2.5 mm | Basic |
| Type BF | 3000 Vac | 5 mm | Double |
| Type CF | 4000 Vac | 8 mm | Double |
3rd Edition - Means of Protection
The 3rd edition of IEC 60601-1 extends the patient focus to require an overall means of protection (MOP) that combines one or more “means of operator protection” (MOOP) and “means of patient protection” (MOPP). So, while the basic provisions of the 2nd and 3rd editions to guard against failure remain, the 3rd edition recognizes that the potential hazards seen by each user can be quite different; an operator has access to a control panel, for example, while the patient may be “connected” via probes.
For 2nd edition Type B use, the standard requires a device to have two isolation barriers to meet MOP requirements. However, a non-medical rated supply may be satisfactory as long as it has reduced leakage currents below 500 µA – this meets what the 3rd edition refers to as “one MOPP” protection. 2nd edition Type BF applications need to meet the 3rd edition’s “two MOOP” and “one MOPP” requirements, and 2nd edition Type CF not only requires an IEC 60601-1 qualified supply, but must ensure an additional isolation barrier between the supply and the part that touches the patient. The latter mandate is typically met with an isolation transformer or a dc-dc converter with 8 mm creepage and double insulation - per the “two MOPP” classification shown in figure 3.
| 3rd Edition Requirements by Classification | |||
|---|---|---|---|
| Classification | Isolation | Creepage | Insulation |
| One MOOP | 1500 Vac | 2.5 mm | Basic |
| Two MOOP | 3000 Vac | 5 mm | Double |
| One MOPP | 1500 Vac | 4 mm | Basic |
| Two MOPP | 4000 Vac | 8 mm | Double |
It is important to note that the changes from 2nd to 3rd edition are more a matter of definition than performance. Although determining whether a particular circuit or function falls under MOOP or MOPP categories can be difficult, a power supply that only incorporates MOOP is generally less expensive than one that also includes MOPP. However, adding MOPP later would be costly, often making it best to go with a supply that meets both criteria.
3rd Edition – Analyzing Risk
The above distinctions are quite specific, but the broader principle underlying revisions to the standard is one of understanding safety and managing risk. This is why the title of the 3rd edition uses the phrase “basic safety and essential performance” in place of “basic safety,” as used by the 2nd edition. Avoiding unacceptable risk in the event of failure or degraded performance or functionality is a prime objective of the 3rd edition. Hence, it requires equipment developers to establish a risk management process, determine acceptable levels of risk, and show that the remaining risk is acceptable.
The 3rd edition standard specifically calls out the Risk Management Process described in ISO 14971 that includes a risk management file where identifiable fault conditions are identified and assessed.
It also demands greater interaction between the equipment manufacturer and the test laboratory, as illustrated in figure 4. This distinguishes between the criteria established by the risk analysis and the test processes used to demonstrate compliance.
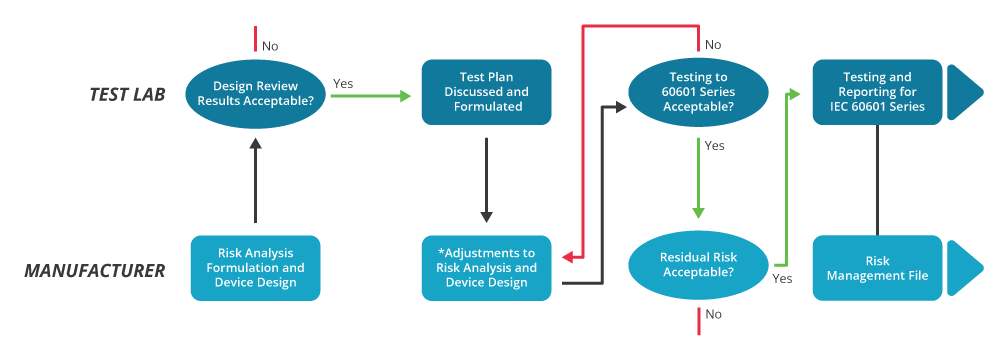
Edition 3.1 – Addressing 3rd Edition Ambiguities
60601-1 Edition 3.1 was introduced in 2012 by the IEC to address many issues identified as unclear or ambiguous in the original 3.0 standard that was released in 2005. Formally referred to as IEC 60601-1: 2005+AMDI: 2012, this updated standard includes almost 500 changes and clarifications across a spectrum of subjects, including essential performance, risk management, mechanical testing, temperature testing, and humidity testing. The amended standard also defines several new specifications for mechanical and electrical hazards.
4th Edition – Addressing EMC Concerns
IEC 60601-1-2 4th edition expands on the risk analysis approach developed previously by delving more deeply into what we might simply call “EMC concerns.” The new IEC 60601-1-2 collateral standard describes this as, “The basic safety and essential performance of medical equipment and systems in the presence of electromagnetic disturbances and electromagnetic disturbances emitted by that equipment and systems.”
In other words, IEC 60601-1-2 4th edition addresses a reciprocal concern about EMC affecting both the medical device in question and other devices in the vicinity. The purpose of this additional risk analysis is to determine appropriate levels of electromagnetic immunity, taking account of the equipment’s intended operating environment and anticipated levels of disturbances.
Part of the reason for introducing this requirement lies in the growing use of devices like smart phones, laptops, tablets, etc. that communicate wirelessly using a cellular network, Wi-Fi, Bluetooth and other wireless protocols.
In the past, it may have been possible to exclude these sources of interference from sensitive medical environments, such as hospitals, but with more and more medical equipment now intended to operate in home and other environments, that approach is no longer viable.
4th Edition – Intended use Environments
Instead, IEC 60601-1-2 4th edition considers immunity in terms of “intended use environments” and no longer includes the “life-supporting” equipment category, a definition which led to uncertainty about whether a particular piece of equipment needed to comply with those requirements or not. Specifically, the intended use environments are:



Complying with the EMC requirements detailed in the 4th edition requires a number of tests as defined by other standards, including more rigorously defined ac supply conditions. The differences between the 3rd and 4th edition of the IEC 60601-1-2 standard are quite extensive, and hence, are detailed in appendix A of this white paper.
THE GLOBAL ADOPTION OF IEC 60601-1
With the United States, Canada, and Europe now having fully adopted the 4th edition EMC requirements, manufacturers intending to ship products to these regions must ensure their products comply to the latest standard. However, the same is equally true for manufacturers in territories that have yet to transition from previous editions of the standard. While transition dates remain unclear in various regions around the world, there is still incentive for manufacturers to stay on top of global standards to avoid being caught out by varying regional compliance.
Figure 5 on the following page illustrates the adoption timeline of 60601-1 as it relates to various territories around the globe. The following sections provide further detail.
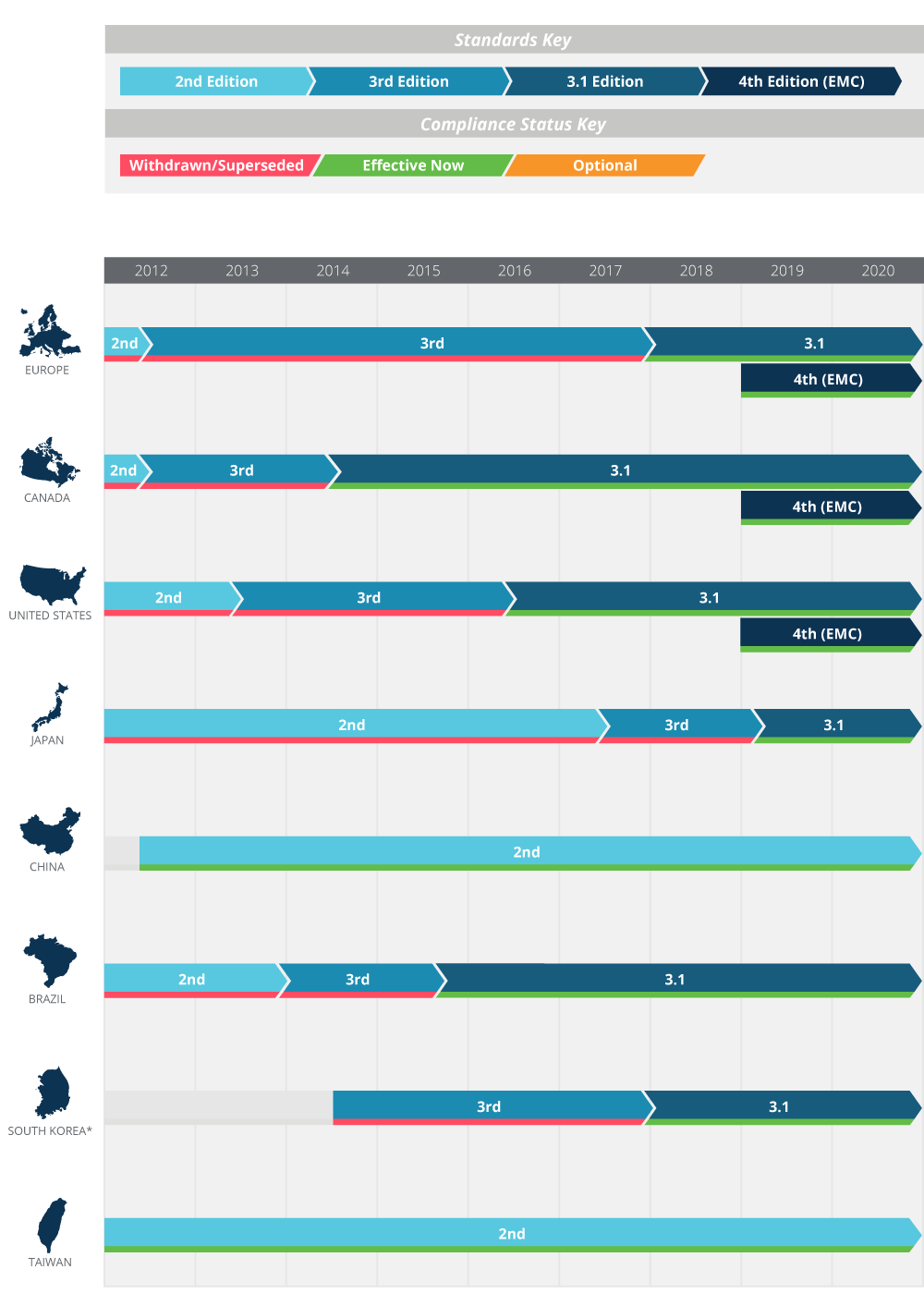
Figure 5: Regional compliance requirements. See appendix B for additional details

Europe
Member countries of the European Union and many other countries in Europe now comply with Edition 3.1 (EN 60601-1 A1:2013), with adoption of the 4th edition EMC standard (EN 60601-1-2:2015) having begun on December 31, 2018. The withdrawn 3rd edition of the standard, known in Europe as EN60601-1:2006, previously superseded the 2nd edition on June 1, 2012, but was then phased out in favor of Edition 3.1 on December 31, 2017.
North America
The US and Canada currently require compliance with Edition 3.1 and originally targeted April 1, 2017 for compliance with the changes required by IEC 60601-1-2. This was subsequently amended to December 31, 2018 to harmonize with the EU timeline for its adoption of the 4th edition EMC standard. Both new products and pre-existing products that are subject to a design change must now comply with the 4th edition EMC standard as of January 1, 2019.

The Rest of the World
Adoption of the various revisions of IEC 60601-1 in other parts of the world is a mixed picture, with transition to the 4th edition EMC standard being unclear:





Because of this unclear picture, equipment intended to ship internationally may require designs that can either meet more than one revision of the standard or multiple versions of the same design, all of which entails additional effort.
SUMMARY
The IEC 60601-1 standard is complex, and has become ever more complicated based on its many regional variations. The related collateral and particular standards often present additional requirements while some simply provide guidance. Knowing which you need to follow and be formally certified as complying with can be challenging, as there are many areas of conflict, confusion, and ambiguity. The lengthy timelines associated with rolling out revisions further complicates matters.
Simply shipping a medical device with basic documentation to a certification lab is no longer adequate. Comprehensive, carefully structured documentation is needed for the design analysis, the design process, and the design rationale with explanations for why certain elements were or were not included or undertaken. The increasing emphasis on risk assessment and considerations about use environments adds to this burden.
In order to help ease the compliance process for medical device designers, CUI offers a comprehensive line of external and embedded medical power supplies ranging from 6 watts to 550 watts that have been certified to IEC 60601 Edition 3.1 two MOPP safety standards and 4th edition EMC standards.
APPENDIX A
Comparison Between 3rd and 4th Edition immunity Test Requirements
| Ac Test Voltage and Power Frequency | ||
|---|---|---|
| Test | 3rd Edition | 4th Edition |
| CISPR 11: Ac Emissions | Nominal | Any voltage of frequency |
| CISPR 11: Radiated Emissions | Nominal | Any voltage of frequency |
| IEC 61000-3-2: Harmonics | 230 Vac/50 Hz (sometimes 100 Vac/50 Hz and 100 V 60 Hz) or rated voltage | Rated single voltage or 230 Vac for single-phase 400 Vac for 3-phase, 50 or 60 Hz |
| IEC 61000-3-3: Voltage Fluctuations and Flicker | 230 Vac/50 Hz or rated voltage | Rated single voltage or 230 Vac for single-phase 400 Vac for 3-phase, 50 or 60 Hz |
| IEC 61000-4-2: Electrostatic Discharge | Any voltage or frequency | Any voltage or frequency |
| IEC 61000-4-3: Radiated RF immunity | Any voltage or frequency | Any voltage or frequency |
| IEC 61000-4-4: Electrical Fast Transients/Burst | Lowest and highest rated voltage, any frequency | Any voltage or frequency |
| IEC 61000-4-5: Surge | Lowest and highest rated voltage, any frequency | Any voltage or frequency |
| IEC 61000-4-6: Conducted RF Immunity | Any voltage or frequency | Any voltage or frequency |
| IEC 61000-4-8: Power Frequency Magnetics | Any voltage, 50 and 60 Hz | Any voltage, 50 and 60 Hz |
| IEC 61000-4-11: Voltage Dips, Dropouts & interruptions | Min and max rated voltage, min power frequency | If rated voltage range <25% of lowest rated voltage, any one voltage. Otherwise, lowest and highest rated voltage |
| IEC 61000-4-2: Electrostatic Discharge | ||
|---|---|---|
| Test | 3rd Edition | 4th Edition |
| Contact Discharges | ±2, 4, 6 kV | ±2, 4, 8 kV |
| Air Discharges | ±2, 4, 8 kV | ±2, 4, 8, 15 kV |
| IEC 61000-4-3: Radiated RF Immunity | ||
|---|---|---|
| Test | 3rd Edition | 4th Edition |
| Enclosures | 3 V/m, Life Support: 10 V/m 80% AM at 1 kHz or 2 Hz, 80 ~ 2500 MHz | 3 V/m Home: 10 V/m 80% AM at 1 kHz or risk frequency 8 ~ 2700 MHz |
| IEC 61000-4-4: Electrical Fast Transients | ||
|---|---|---|
| Test | 3rd Edition | 4th Edition |
| Ac Mains or Dc Input | ±2 kV, 5 hHz PRF | ±2 kV, 100 kHz PRF |
| Input/Output Ports | ±1 kV, 5 kHz PRF | ±1 kV, 100 kHz PRF |
| IEC 61000-4-5: Surges | ||
|---|---|---|
| Test | 3rd Edition | 4th Edition |
| Ac Mains, Line to Ground | ±0.5, 1, 2 kV | ±0.5, 1, 2 kV |
| Ac Mains, Line to Line | ±0.5, 1 kV | ±0.5, 1 kV |
| Dc Input (>3 m), Line to Ground | N/A | ±0.5, 1, 2 kV |
| Dc Input (>3 m), Line to Line | N/A | ±0.5, 1 kV |
| Input/Output, Line to Ground | N/A | ±2 kV (outdoor lines only) |
| IEC 61000-4-6: Conducted RF Immunity | ||
|---|---|---|
| Test | 3rd Edition | 4th Edition |
| Ac Mains | 3 V Life Support: 10 V ISM 1 kHz or 2 kHz 80% AM 150 kHz ~ 80 MHz |
3 V with 6 V ISM. Home: 6 V Amateur 80% AM at 1 kHz or risk frequency 150 kHz ~ 80 MHz |
| Dc & I/O Patient Couples (>3 m) | 3 V Life Support: 10 V ISM 1 kHz or 2 kHz 80% AM 150 kHz ~ 80 MHz |
3 V with 6 V ISM. Home: 6 V Amateur 80% AM at 1 kHz or risk frequency 150 kHz ~ 80 MHz |
| IEC 61000-4-8: Power Frequency Magnetics | ||
|---|---|---|
| Test | 3rd Edition | 4th Edition |
| Enclosures | 3 A/m, 50 and 60 Hz | 30 A/m, 50 and 60 Hz |
| IEC 61000-4-11: Voltage Dips, Dropouts, & Interruptions | ||
|---|---|---|
| Test | 3rd Edition | 4th Edition |
| Voltage Dips (16 A) | >95% dip, 0.5 periods 0° and 180° 60% dip, 5 periods 30% dip, 25 periods |
100% drop, 0/5 periods, 0°, 45°, 90°, 180°. 225°. 270°, 315° 100% dip, 1 period 30% dip, 25/30 periods |
| Voltage Interruptions (all input current) | >95% drop, 5 sec | 100% drop, 5 sec |
| IEC 61000-4-3: Rated RF Immunity & RF Wireless Communications Equipment | |||||
|---|---|---|---|---|---|
| Test (MHz) | Band (MHz) | Service | Modulation | Max Power (W) | Test Level (V/m) |
| 385 | 380-390 | TETRA 400 | 18 Hz pulse | 1.8 | 27 |
| 450 | 430-470 | GMRS 460, FRS 460 | FM ±5 kHz 1 kHz sine |
2 | 28 |
| 710 | 704-787 | LTE Band 13, 17 | 217 hz pulse | 0.2 | 9 |
| 745 | |||||
| 780 | |||||
| 810 | 800-960 | GSM 800/900; TETRA 800; iDEN 820; CDMA 850; LTE Band 5 | 18 Hz pulse | 2 | 28 |
| 870 | |||||
| 930 | |||||
| 1720 | 1700-1990 | GSM 1800; CDMA 1900; GSM 1900; DECT; LTE Band 1, 3, 4, 25; UMTS | 217 Hz pulse | 2 | 28 |
| 1845 | |||||
| 1970 | |||||
| 2450 | 2400-2570 | Bluetooth; WLAN; 802.11 b/g/n; RFID 2450; LTE Band 7 | 217 Hz pulse | 2 | 28 |
| 5240 | 5100-5800 | WLAN 802.11 a/n | 217 Hz pulse | 0.2 | 9 |
| 5500 | |||||
| 5785 | |||||
APPENDIX B
| Regional Compliance by Country | |||
|---|---|---|---|
| Region/Country | Standard | Revision Timeline | Compliance Status |
| Europe | 2nd Edition | prior to June 1st, 2012 | Withdrawn |
| 3rd Edition | since June 1st, 2012 | Superseded | |
| Edition 3.1 | from January 1st, 2018 | Effective Now | |
| 4th Edition (EMC) | from January 1st, 2019 | Effective Now | |
| Canada | 2nd Edition | prior to June 1st, 2012 | Withdrawn |
| 3rd Edition | since June 1st, 2012 | Superseded | |
| Edition 3.1 | since August 31st, 2014 | Effective Now | |
| 4th Edition (EMC) | from January 1st, 2019 | Effective Now | |
| United States | 2nd Edition | prior to July 1st, 2013 | Withdrawn |
| 3rd Edition | since July 1st, 2013 | Superseded | |
| Edition 3.1 | since August 31st, 2016 | Effective Now | |
| 4th Edition (EMC) | from January 1st, 2019 | Effective Now | |
| Japan | 2nd Edition | prior to June 1st, 2017 | Withdrawn |
| 3rd Edition | since June 1st, 2017 | Superseded | |
| Edition 3.1 | from March 1st, 2019 | Effective Now | |
| 4th Edition (EMC) | To be Determined | Optional | |
| China | 2nd Edition | since June 1st, 2012 | Effective Now |
| 3rd Edition | To be Determined | Optional | |
| Edition 3.1 | To be Determined | Optional | |
| 4th Edition (EMC) | To be Determined | Optional | |
| Brazil | 2nd Edition | prior to January 1st, 2014 | Withdrawn |
| 3rd Edition | since January 1st, 2014 | Superseded | |
| Edition 3.1 | since September 25th, 2015 | Effective Now | |
| 4th Edition (EMC) | To be Determined | Optional | |
| South Korea | 3rd Edition | since June 1st, 2014 | Superseded |
| Edition 3.1 | from January 1st, 2018 | Effective Now | |
| 4th Edition (EMC) | To be Determined | Optional | |
| Taiwan | 2nd Edition | - | Effective Now |
| 3rd Edition | To be Determined | Optional | |
| Edition 3.1 | To be Determined | Optional | |
